

Research
Key questions of our research are:
• What controls cell-to-cell heterogeneity within a population of pathogens?
• What are the biochemical or molecular factors that trigger a switch in antigen expression?
• How does ncRNA contribute to the establishment of distinct interchromosomal DNA-DNA interactions?
• Can RNA-processing hotspots serve as post-transcriptional enhancers?
Technologies used by our group include:
• Genome-wide chromosome conformation capture (Hi-C and Micro-C) to study the 3D genome architecture
• RADICL-seq to identify regulatory, trans-acting RNA molecules
• Ribosome-profiling to study translational control and to identify ncRNA
• ChIP- and Apex2-based methods to study proteins and RNA associated with specific genomic loci
• Single-cell RNA-seq, lineage-tracing and scSLAM-seq (in collaboration with Florian Erhard, University of Würzburg) to yield temporal omic information on molecular triggers of cell-to-cell heterogeneity
Why study antigenic variaton?
Antigenic variation is of high medical relevance and represents an excellent system to study the mechanisms controlling cell-to-cell heterogeneity across scales, i.e. at the biochemical, molecular, cellular and population level.
Antigenic variation refers to the capacity of an infecting organism to systematically alter the identity of proteins displayed to the host immune system making it difficult or impossible for the host to eliminate the pathogen. Successful antigenic variation requires mechanisms ensuring the expression of a single antigen from a family of genes coding for different antigen isoforms and a mechanism ensuring the periodic switching from the expression of one antigen isoform to another.
Decades of research have made the unicellular parasite Trypanosoma brucei one of the most important models for antigenic variation research. In addition, the extremely high expression of a single antigen makes the parasite an ideal organism to monitor changes in antigen expression using single-cell omics approaches.
Over the last several years, combining Hi-C, ATAC-seq and single-cell RNA-seq, my group has been able to show that the deletion of specific histone variants can affect the 3D genome architecture and leads to increased ant
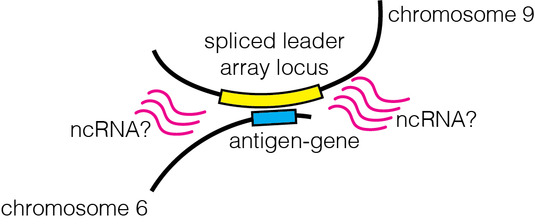
igen switching in T. brucei (Müller et al., Nature 2018). Furthermore, we found that the gene coding for the active antigen makes frequent DNA-DNA interactions with a locus important for RNA maturation
(Faria et al., Nature Microbiology 2021). These findings point to the existence of posttranscriptional, enhancer-like mechanisms in an organism where classic promoter-enhancer interactions are absent. Going forward, we hope to identify the factors, possibly ncRNA, that contribute to the establishment of the observed 3D genome architecture.
In summary, I believe that the recent development of single-cell omics technologies and Cas-based expression-perturbation and genome editing tools has created unprecedented opportunities to use antigenic variation as a model to elucidate the mechanisms controlling cell-to-cell heterogeneity.
Biology at the single cell level: antigenic variation and beyond
Having established numerous omics approaches in different unicellular pathogens, my long-term goal is to bring these technologies to the single-cell level and to be able to perform multiple analyses on the same cell. In the future, we hope to be able to take an image of a cell, determine its genome sequence, transcriptome and proteome and repeat this for thousands of cells. Furthermore, we hope to develop temporal omics approaches that will allow us to link molecular events over time. Such analyses will allow us to identify factors responsible for a switch in antigen expression and help to reveal the mechanisms controlling the level of heterogeneity in pathogen populations.
To fund this research, I have just been awarded an ERC-CoG. In addition, together with a colleague at the University of Gießen, I have established an EU-funded integrative training network (ITN) focusing on cell-to-cell heterogeneity in unicellular organisms (www.cell2cell.eu). The goal of the network is to bring together experts in chromatin and RNA biology; in single-cell isolation and analysis; in live-cell imaging and super-resolution microscopy; and in bioinformatics and mathematical modelling. The members of the network are from academia and industry and are located at Helmholtz Institute for Environmental Health, Munich; Institut Curie, Paris; Karolinska Institutet, Stockholm; Swiss Tropical and Public Health Institute, Basel; Weizmann Institute, Tel Aviv; IMM, Lisbon; University of Oxford; Radboud University, Nijmegen; University of San Francisco; University of Tokyo; CellSorter Inc., Budapest; Epigenetiks Inc., Istanbul; CYBP Inc., Tokyo. Together, we hope to develop technologies for the study of unicellular organisms at the single cell level.